| главная | архив | ссылки | студентам | программы | о проекте | |
Патогенез головной боли напряжения Проблема головной боли развивается параллельно с изучением патофизиологических механизмов боли. До сих пор окончательно не решен вопрос о том, как формируется в организме то ощущение, которое интерпретируется как боль. Важный шаг в изучении этого был сделан Melzak и Wall в 1965г., которые открыли в ЦНС систему интернейронов желатинозной субстанции, контролирующую прохождение болевых импульсов. Этой системе было дано название - "система воротного контроля боли". В том же году H. Wolff экспериментально выделил активное вещество - плазмокинин или субстанцию Р, которая играет значительную роль в формировании болевого ощущения. Что же может болеть в голове? На этот вопрос Р. Боконжич (1984) дает следующий ответ: "Болеть могут те структуры, которые в большей или меньшей степени имеют чувствительные рецепторы. Они, как и в других участках тела, располагаются с неодинаковой частотой в различных областях головы". Боль в области головы воспринимается болевыми рецепторами, расположенными в твердой мозговой оболочке на основании черепа, синусах, крупных артериях мозга на его основании и конвекситальной поверхности, менингеальных артериях, дупликатуре твердой мозговой оболочки в области серпа мозга и намета мозжечка. Не реагируют болью: твердая мозговая оболочка на конвекситальной поверхности, большинство отделов мягкой и паутинной мозговых оболочек, эпендима хориоидальных сплетений, остальные кровеносные сосуды и черепные нервы, большинство участков паренхимы мозга. Внечерепные рецепторы, способные генерировать болевые сигналы сосредоточены в коже, подкожных тканях, мышцах, сухожилиях, апоневрозах, артериях, зубах, полости рта и носа, мышцах носа и среднего уха. Не реагируют болью поверхностные вены, кости черепа и диплоэ. В проведении болевых импульсов от рецепторов ЦНС участвуют краниальные чувствительные нервы: Y,YII,IX,X и первые три шейных корешка спинного мозга. Остается не до конца раскрытым аспект проецирования болей. Вероятно, это связано с многочисленными причинами, влияющими на формирование паттерна ГБ. Следует четко понимать, что ГБ существенно отличается от различных вариантов прозопалгий менее выраженным, недостаточно дифференцированным ноцицептивным ощущением, более обобщенным болевым рисунком и ярким сосудисто-вегетативным сопровождением. Учитывая это, имеются весьма веские основания говорить об участии в формировании ГБ преимущественно "дорсального пути спинномозгового тракта", который входит в палиоспинноталамическую систему, и направляясь к ноцицептивным структурам ретикулярной формации ствола мозга и вентро-медильным ядрам таламуса, является основным каналом передачи плохо локализованной ноцицептивной информации. Первые попытки научного объяснения патогенеза и патофизиологии головной боли напряжения принадлежат Wolff (1963). Эксперименты, проведенные на больных и здоровых людях, позволили ему создать модель развития ГБ этого типа. Причиной ГБН он считал длительное напряжение скелетных мышц головы и шеи, являющееся следствием тревожности пациента. Длительный спазм скелетных мышц головы и шеи проявляется болью в форме чувства сдавления, стягивания, ощущения шлема и др. Повышение напряжения мышц приводит к сужению крупных артериальных сосудов, появлению ишемии и усилению неприятных ощущений. Нарушение кровотока сопровождается венозным застоем и, таким образом, возникает порочный круг. Мышца недостаточно снабжается кровью, а вследствие усиления напряжения в ней накапливаются продукты метаболизма, которые не могут быть в соответствующей степени выведены через венозную сеть, при этом мышца становится отечной и болезненной. С помощью метода электромиографии Wolff доказал, что мышечное напряжение наиболее выражено в тех мышцах головы, где локализуется головная боль. Вслед за этим, Hopkins и Ziegler (1988) установили, что потенциалы действия мышц скальпа выше при мигрени, чем при ГБН, следовательно, этот признак никак нельзя было считать патогномоничным для ГБН. Gerber обнаружил, что у части пациентов с ГБН напряжение экстракраниальной мускулатуры не выявляется вовсе. У другой же группы больных оно, очевидно, является реакцией на стресс или следствием интенсивности самой ГБ. Ряд исследователей предполагают, что повышенная концентрация калия, возникающая во время длительного напряжения скелетных мышц головы и шеи, стимулирует хеморецепторы и вызывает ГБ. При дополнительной ишемии (сдавление сонных артерий или применение вазоконстрикторов) боль усиливается, что указывает на ее ишемическую природу. Кроме этого, у больных возникает спонтанное снижение пульсового кровотока, т.е. интракраниальные артерии находятся в состоянии сужения. Р.Боконжич (1984) выделяет 2 параллельных фактора, вызывающих головную боль при напряжении мышц скальпа: напряжение самих мышц, ишемия, отек и химические изменения в них; конкурентное сужение артерий, еще более усугубляющее ситуацию. В.Н.Шток (1988) выдвигает 2 возможных механизма возникновения ГБН. Первый - это активация передачи импульса в нервно-мышечном синапсе (генерализованные нейрогуморальные сдвиги); второй- активация мышечного напряжения по сегментарному механизму (раздражающее действие местных факторов). Pfaffenrath и Gerber (1992) предполагают, что при ГБН имеет место функциональный отказ защищающей от боли антиноцицептивной системы. Биогенные амины, такие как серотонин, норадреналин и допамин, а также эндорфины и нейроэндокринные факторы играют в этой системе ведущую роль. У пациентов с ГБН может иметь место недостаточное (нефизиологичное) реагирование вазомоторной системы на раздражение в перманентной стрессовой ситуации. А.М.Вейн (1994) пишет: "Не вызывает сомнений, что в основе ГБН существенную роль играет наличие хронического эмоционального стресса, который формируется под влиянием индивидуально значимых психогенных факторов у лиц с определенными особенностями личности и недостаточностью механизмов психологической защиты, а также функциональной недостаточностью антиноцицептивных систем. Указанные нарушения приводят к возникновению вегетативно-эндокринной и психомоторной активации, что проявляется повышением мышечного тонуса, ишемией, отеком и биохимическими проявлениями в мышечной ткани" (схема 1). 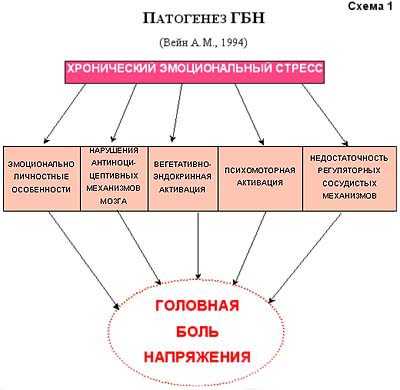 Концепция А.М. Вейна получила дальнейшее развитие в работах других отечественных исследователей. Согласно данным Е.Я. Страчунской (1996), основными факторами формирования ГБН являются невротические особенности личности и наличие хронического стресса, что приводит к нарушению функционального состояния лимбико-ретикулярного комплекса. Это сопровождается изменениями в действии ноци- и антиноцицептивной систем, развитием тревожно-депрессивного синдрома и нарушением функционального состояния системы "тройничный-лицевой нервы". В результате возникает гипертонус и гипоксия перикраниальных и мимических мышц, что выражается клиническим проявлением ГБ. 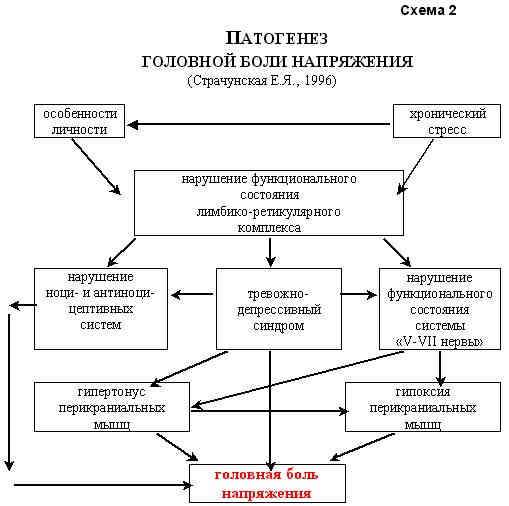 Т.о., патофизиологические основы ГБН продолжают активно изучаться. Уже сегодня не вызывает сомнений тот факт, что в возникновении и оформлении этого патологического состояния играют определенную роль врожденные особенности личности, на которые наслаивается переживание текущих стрессовых ситуаций. ГБН оформляется и прогрессирует, переходя из эпизодического в хронический вариант у лиц с хроническим стрессом, имеющим определенную дефектность антиноцицептивных механизмов мозга. Длительное применение анальгетиков осложняет течение заболевания, приводя к абузусному синдрому. Однако отдельные конкретные механизмы патогенеза ГБН остаются недостаточно изученными. В этой связи, следует остановиться на психологических аспектах ГБН. Еще Wolff (1963) установил, что головная боль является только частью реакции индивидуума на жизненные трудности. Кроме нее, имеют место ощущение утомления, напряженности и депрессивное состояние. Вместе с тем нередко отмечается наличие обсессивно - компульсивных невротических реакций. Больной обычно является сенситивной, обязательной личностью, хронически психически напряжен, у него отмечается специфическая тревожность, сопровождающаяся страхом перед возможным обнаружением скрытой ненависти и непереносимости по отношению к родителям или близким людям. В эксперименте на людях Wolff показал, что повышенная концентрация внимания у студентов и определенная степень предэкзаменационной тревоги провоцируют напряжение поперечно-полосатых мышц лба, головы и шеи, вазоконстрикцию экстракраниальных сосудов и появление боли. Friedman (1961) предполагал, что ГБН находится в тесной связи с временным эмоциональным конфликтом, который осознан только частично. Fine (1969) в своей обширной статье излагал психоаналитическую концепцию об особенностях личности больных, страдающих ГБ. Пациенты часто находятся в конкурентных отношениях с теми, кого они любят и ненавидят, имеют специфическую фантазию и подсознательное желание разрушить вместилище своего интеллекта и успеха своих конкурентных переживаний - мозг и голову, как представительство их психических возможностей. Это желание оборачивается против себя самого, как "казнь", "наказание" за запрещенное желание. Gittleson (1962) в своей статье о локализации ego у лиц с психогенной ГБ пришел к выводу, что большинство из них оценивают голову как наиважнейшую часть тела и рассматривают ее как физическое вместилище ego. Martin, Fine (1967) считали, что причиной хронического ощущения тревоги у больных с ГБ является страх перед ощутимым запрещенным чувством ненависти и неприемлемыми сексуальными порывами. На основании этого можно утверждать, что ГБН является физическим выражением чувства хронической тревоги, в генезе которой лежит ощущение вины, стыда, беспомощности и брошенности. Между тем ощущение обиды или, даже, ненависти к родителям или любимым людям значительно угнетено. Когда это ощущение вытесняется, тревога становится трудно сдерживаемой, что приводит к проявлению ее физического симптома - напряжению мышц головы и шеи. Клинические наблюдения достаточно убедительно свидетельствуют о том, что сложная жизненная ситуация определяет возникновение невротического состояния, его же тип и форма во многом зависят от достаточно стабильного личностно-типологического своеобразия человека. По мнению О.А. Колосовой и Е.Я. Страчунской (1995), больные с ГБН - это экстравертированные личности с ярко выраженной тревожностью и относительно кратким анамнезом жизненных проблем. При наличии у них хронической формы заболевания, эти пациенты обычно становятся апатичными или депрессивными. Они без конца жалуются на ГБ, но весьма скудно ее описывают, что затрудняет выяснение деталей заболевания. Изложенные данные, позволяют обосновать тот факт, что в основе возникновения ГБН существенную роль играет наличие хронического эмоционального стресса, который формируется под влиянием индивидуально значимых психогенных факторов у людей с определенными личностными особенностями, а также функциональной недостаточностью антиноцицептивных систем. Указанные нарушения приводят к вегетативно-эндокринной и психомоторной активации, что проявляется повышением мышечного тонуса, ишемией, отеком и биохимическими изменениями в перикраниальных тканях. Возможные патофизиологические механизмы тревоги В психологическом отношении тревога является ожиданием неизвестной отрицательной перспективы. Как эмоциональный феномен, тревога представляет собой отрицательно окрашенное ожидание несчастья или опасности. Несомненно, тревога имеет эволюционно-приспособительное значение в организации защиты и избегания опасности. Создание в 1960-1962гг. бензодиазепиновых транквилизаторов (БДТ), оказывающих выраженное противотревожное действие, дало возможность приблизиться к расшифровке патофизиологических механизмов тревоги. Экспериментально было показано, что противотревожный эффект БДТ (наряду с их противосудорожным и миорелаксирующим свойствами) обусловлен усилением функции тех синапсов мозга, где роль медиатора выполняет -аминомаслянная кислота, (так называемые ГАМК-ергические синапсы). В синапсах мозга ГАМК является медиатором торможения. Воздействуя на белковый субстрат мембран нервных клеток, называемый рецепторами ГАМК, медиатор изменяет структуру белковых молекул этих рецепторов и открывает управляемые ими хлорные каналы. В результате этого возрастает проницаемость мембран нервных клеток для ионов Cl-, возникает тормозящий постсинаптический потенциал, и чувствительность нервных клеток к возбуждающим стимулам снижается. На основе экспериментальных данных, появилась возможность рассматривать тревогу как состояние, в основе которого лежат дефицит ГАМК-ергического торможения, повышенная возбудимость и избыточная импульсная активность некоторых структур головного мозга. Предполагалось, что дефицит ГАМК-ергического торможения является результатом воздействия образующихся в мозговой ткани эндогенных модуляторов-ингибиторов бензодиазепиновых рецепторов (БДР). Это предположение опиралось на факты, свидетельствующие о существовании белковых пептидных и небелковых соединений, преимущественно производных B-карболина, которые также специфически связываются с БДР, но противодействуют анксиолитическому действию бензодиазепиновых препаратов, ослабляют или устраняют влияние ГАМК на нейроны мозга и оказывают анксиогенное действие на человека. Препятствуя взаимодействию образующихся в мозге анксиогенных веществ с БДР, бензодиазепиновые транквилизаторы усиливают ГАМК-ергическое торможение импульсной активности нейронов, прежде всего коры полушарий мозга, миндалевидного и септикогиппокампального комплексов. Идея о ГАМК-ергической природе тревожных состояний основывалась, во-первых, на известных представлениях о тормозящей функции ГАМК в мозгу, и, во-вторых, на допущении возможности на этой основе интерпретировать анксиолитическое действие не только бензодиазепиновых, но и небензодиазепиновых транквилизаторов: барбитуратов и алкоголя, давно используемых при состояниях тревоги и, как оказалось, способных усиливать ГАМК-ергическое торможение и эффекты ГАМК, ряда производных В-карболина. При анализе клинических данных возникают сомнения в справедливости представлений об исключительной роли ГАМК-ергических механизмов в генезе тревоги. По-видимому, разнообразие проявлений и причин тревожно-анксиозных нарушений объясняется неоднородностью их патофизиологических механизмов. О том же свидетельствует неэффективность однонаправленного лечения тревоги разного происхождения (тревога в предделириозном состоянии, в рамках галлюцинаторно-параноидного симптомокомплекса, тревога невротического регистра и т.д.). Различные факты свидетельствуют о том, что анксиолитический феномен может возникать при вовлечении разных нейронных или нейрохимических "мишеней". Следовательно, ГАМК-ергический механизм не является, по крайней мере, единственным в генезе тревоги. Экспериментально была показана существенная роль серотонина в инициации тревожных расстройств. Представления о важной роли серотонина в регуляции уровня тревожности опираются на убедительные факты: серотонинсодержащие нейроны, тела которых расположены в ядрах шва среднего мозга (ЯШСМ), дают начало восходящей системе аксонов, образующих два пути: 1) от клеток дорсального ядра преимущественно к структурам полосатого тела; и 2) от нейронов медиального ядра к структурам Hpth, септикогиппокампальной области и коре полушарий мозга. Присущая серотонину способность угнетать импульсную активность нейронов ЯШСМ лежит в основе механизма их самоингибирования. При возбуждении нейронов ЯШСМ серотонин высвобождается не только в синапсах, образованных аксонами этих клеток на нейронах переднего мозга, но и в синапсах, образованных сомой серотонинергических нейронов. Высвобожденный в дендросоматических синапсах серотонин, активируя локализованные здесь рецепторы серотонина (Р1а-СТ), аутоингибирует импульсную активность нейронов ЯШСМ. Этим объясняется, почему микроионофоретическое подведение серотонина к телам нейронов ЯШСМ подавляет импульсную активность большинства серотонинергических нейронов. Аналогичным действием обладают буспирон и изапирон. Как имитаторы эффектов серотонина, эти препараты отличаются от него лишь лучшей проницаемостью в мозг и большей избирательностью действия на Р1а-СТ рецепторы. По способности связываться с последними они уступают серотонину всего в 10-40 раз, но способность связываться с серотониновыми рецепторами (Р1в-СТ и Р2-СТ) другой локализации в 150-1000 раз слабее выражена, чем у серотонина. Таким образом, анксиолитический эффект буспирона и изапирона является результатом избирательного угнетения этими веществами функции серотонин- ергических нейронов ЯШСМ. Такой механизм анксиолитического действия, полное отсутствие влияния на ГАМК- ергические синапсы и БДР и клинически вполне определенное противотревожное действие буспирона дают основания предполагать, что избыточно повышенная функция серотонинергической системы мозга является патогенетически значимым механизмом тревоги. Хотя не все факты строго соответствуют серотонинергической гипотезе состояний тревоги, а многие аспекты этой проблемы лежат еще вне сферы исследований, идея о ведущей роли серотонинергической системы мозга в происхождении тревоги и действии анксиолитиков может оказаться весьма плодотворной как для понимания патогенеза этой патологии, так и для создания новых противотревожных ЛС. |
|
| главная | архив | ссылки | студентам | программы | о проекте |